

Небольшие совпадающие участки в аминокислотных последовательностях, стоящие в структуре белка друг за другом, называют тандемными повторами. От последовательности аминокислот зависит то, как он свернется после сборки, а от правильной пространственной формы — то, как он будет работать. Российские математики создали программу поиска таких повторов под названием Method to Search for Highly Divergent Tandem Repeats (MSHDTR). О результатах, которые могут свидетельствовать об образовании многих белков путем множественных тандемных дупликаций, ученые рассказали в статье в International Journal of Molecular Sciences.
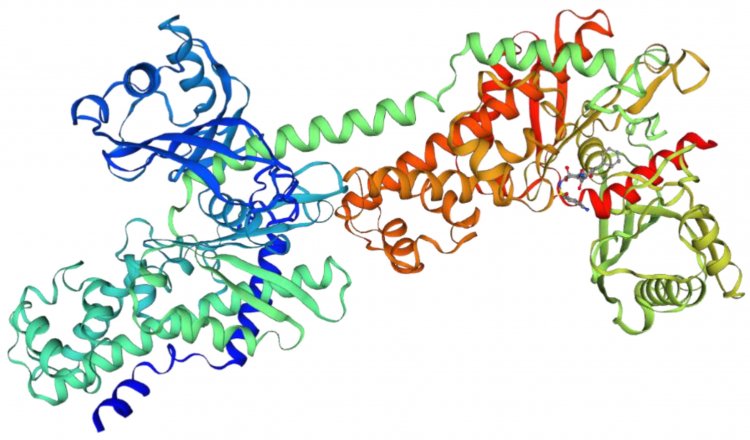
Трехмерная структура гексокиназы-1 из генома Mus musculus, которая содержит тандемный повтор длиной в 64 аминокислоты
Изучение связи между аминокислотными последовательностями и структурой белка — очень важная задача для молекулярной биологии и биоинформатики. Тандемные повторы в аминокислотных последовательностях встречаются в примерно 25% белков и кодируют определенную либо вторичную, либо третичную структуру белка. Длина таких участков может колебаться от двух до нескольких десятков аминокислот. Обычно аминокислотные последовательности с периодом в семь аминокислот сворачиваются в альфа-спирали, а в две аминокислоты — создают бета-слои. Существует много программных продуктов и серверов для обнаружения повторов в аминокислотных последовательностях: одни, как ProStrip, XSTREAM или TRUST, помогают искать их в трехмерных структурах белков, другие, как HHrep или HHRepID, используют скрытые модели Маркова, в основе третьих лежит преобразование Фурье, а алгоритм DAVROS производит расчеты на основании весовой матрицы аминокислот. Они дают довольно точные результаты, предсказывая начало и конец таких повторяющихся аминокислотных мотивов, но плохо определяют повторы с большим количеством вставок и замен аминокислот.
«Эволюционное происхождение новых генов и соответствующих им белков давно интересует исследователей. Одним из способов происхождения генов и белков может быть тандемная дупликация, или удвоение какого-либо фрагмента ДНК. После такого процесса тандемные повторы в белках могут сильно измениться из-за накопления замен аминокислот, их вставок и делеций. Этот процесс эволюционных изменений необходим для улучшения функциональной активности вновь созданного белка. В результате у современных белков периодический мотив во всей либо в значительной части аминокислотной последовательности может быть сильно размытым. Данная работа направлена на поиск таких тандемных мотивов в аминокислотных последовательностях. Мы разработали метод поиска высокодивергентных тандемных повторов, которые могут содержать в среднем до 4,4 замены на одну аминокислоту, тогда как чувствительность всех остальных методов ограничена примерно 2,5 заменами», — рассказал профессор Евгений Коротков, руководитель группы математического анализа последовательностей ДНК и белков ФИЦ Биотехнологии РАН.
Метод российских математиков, получивший сокращенное название MSHDTR, учитывает пары соседних аминокислотных остатков, образующих связи. Кроме того, он группирует аминокислоты как полярные, неполярные, ароматические, положительно или отрицательно заряженные и представляет последовательности в белке в виде пяти символов (количество групп) вместо 20 (примерное количество видов аминокислот). Такой метод признает за тандемные повторы участки, содержащие аминокислоты из одной группы в нужном порядке. Это сделано потому, что вероятность замены аминокислоты в тандемном повторе на другую аминокислоту той же группы выше, чем на «представительницу» иной группы.
Ученые применили его на базе данных Swiss-Prot, содержащей более 500 тысяч аминокислотных последовательностей, и обнаружили более 15 тысяч новых белков с тандемными повторами, длина большинства из которых достигала 5-7 аминокислотных остатков. 14 тысяч последовательностей оказались высокодивергентными — то есть содержали много замен и вставок и были практически невидимыми для других методов. С этими результатами алгоритм MSHDTR составил весовую матрицу, которую можно использовать для других программ, в том числе как примеры для машинного обучения.
«При помощи преобразования Фурье можно обнаружить всего 1707 тандемных повторов из 15 407, которые способен найти наш метод. Это всего 11%. Однако многие аминокислотные последовательности, особенно в ферментах, полностью состоят из сильно дивергировавших тандемных повторов. Фактически мы нашли следы создания белков из повторов различной длины. Но наш метод имеет и некоторые ограничения: он пропускает последовательности, которые повторяются малое число раз. Поэтому разные программы нужно использовать для разных целей. Если мы ищем высокодивергентные тандемные повторы, присутствующие в больших количествах, то разумнее выбрать MSHDTR, а T-REKS и XSTREAM лучше обнаружат малочисленные тандемные повторы, которые сохранились практически без изменений. Таким образом, чтобы охватить весь спектр потенциальных тандемных повторов, мы рекомендуем использовать T-REKS и XSTREAM в комбинации с MSHDTR», — пояснила соавтор работы Валентина Руденко, научный сотрудник группы математического анализа последовательностей ДНК и белков ФИЦ Биотехнологии РАН.
Информация и фото предоставлены пресс-службой ФИЦ Биотехнологии РАН























{kind=link}