

Двое ученых из Сколтеха представили эффективную нейросетевую модель, которая на основе информации о структуре белков предсказывает, какие их участки взаимодействуют с биологическими молекулами из класса пептидов. Такие предсказания нужны для разработки эффективных и нетоксичных пептидных лекарств, которые смогут точечно влиять на взаимодействия между белками в клетке и тем самым регулировать широкий спектр процессов в организме. Исследование опубликовано в издании Journal of Chemical Information and Modeling.
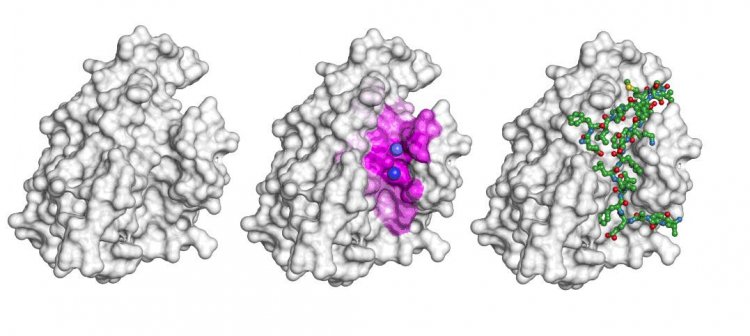
Серая фигура — белок, вступающий во взаимодействие с пептидом (зеленоватый каркас справа). Модель прогнозирует, что взаимодействие локализовано на участке белка, который выделен розовым цветом (по центру), и предсказывает точные сайты связывания — две фиолетовые сферы. Источник: Игорь Козловский и Петр Попов / Сколтех
Белки — рабочие лошадки, которые перемещаются внутри клетки, взаимодействуют друг с другом и обеспечивают своей работой самые разные необходимые организму функции. Для фармкомпаний возможность повлиять на взаимодействия между белками всегда выглядела очень привлекательно. Однако эти потенциальные мишени лекарств представлялись недостижимыми. Ведь крупным лекарственным молекулам так называемых биопрепаратов не достать белки внутри клетки из-за препятствия в виде её оболочки, а низкомолекулярные препараты зачастую не способны оказать на них необходимое воздействие.
Пептиды, которые выступают в роли естественных посредников и регуляторов в 40% клеточных процессов, занимают промежуточное положение между низкомолекулярными и биопрепаратами, объединяя их сильные стороны. С одной стороны, молекулы пептидов достаточно малы, чтобы преодолеть клеточную мембрану и проникнуть в клетку. С другой стороны, для них характерны высокая специфичность и аффинность, то есть они могут воздействовать на белки прицельно и эффективно.
Для разработки пептидных лекарств нужно знать так называемые сайты связывания — участки белковой молекулы, которые вступают во взаимодействие с пептидами. Чем больше сайтов известно, тем шире возможности для создания медицинских препаратов.
Сайты связывания можно обнаружить экспериментальными методами, например, рентгеноструктурным анализом, который позволяет построить трехмерную модель белка в комплексе с биоактивной молекулой, наблюдая дифракцию рентгеновского излучения на его кристалле. Однако такие исследования дороги при переборе большого количества белковых мишеней или биоактивных молекул.
Альтернативой служат вычислительные методы, которые дают результаты быстрее и дешевле. Многие из них предсказывают сайты связывания с использованием машинного обучения. Это значит, что предсказания становятся точнее по мере накопления информации о структуре комплексов «белок — пептид».
В опубликованной 22 июля в Journal of Chemical Information and Modeling работе участники научной группы Сколтеха iMolecule — аспирант Игорь Козловский и старший преподаватель Петр Попов — представили вычислительный метод BiteNetPp, который раскрывает потенциал трехмерных сверточных нейросетей для обнаружения сайтов связывания пептидов с белками. Модель принимает на вход известную структуру белка и выдает предположительные координаты сайтов связывания, а также оценку достоверности своего прогноза.
Как поясняет руководитель исследования Петр Попов, модель обнаруживает сайты связывания по принципу распознавания изображений, который тот же коллектив авторов впервые использовал для задач такого рода в прошлом году: «Нейронные сети уже давно обучают распознавать, например, фигуры пешеходов или велосипедистов на обычных двумерных снимках. Мы рассматриваем обнаружение сайтов связывания как подобного рода поиск объекта на картинке. Разница в том, что мы подаем на вход модели не плоскую картинку, а трехмерную атомарную структуру белка, и модель оперирует не пикселями, а их 3D-аналогом — вокселями».
Представленная в новом исследовании модель расширяет сферу применения прошлогодней. «Это называется доменной адаптацией. BiteNetPp — первая модель, которую дообучили на датасете по белкам и пептидам после первичного обучения на данных по белкам и малым молекулам, — объясняет Попов. — Наглядная аналогия могла бы выглядеть так: вы хотите обучить модель обнаруживать места, где на улице останавливаются велосипедисты, но вы начинаете с данных об остановках пешеходов и лишь затем расширяете домен до велосипедистов. Вместо того чтобы начинать с чистого листа, вы переучиваете готовую модель, ожидая, что „сайты связывания“ велосипедистов и пешеходов могут пересекаться: может, это ларек с мороженым, светофор и т.п.».
Авторы модели сравнили предсказания BiteNetPp и основных используемых сегодня вычислительных методов предсказания сайтов связывания пептидов белком и продемонстрировали, что модель стабильно превосходит конкурирующие методы по точности воспроизведения экспериментальных данных. При этом немаловажно, что модель проводит анализ одной белковой структуры менее чем за секунду, что делает метод применимым в масштабных исследованиях. Существуют тысячи белок-белковых взаимодействий, потенциально подверженных влиянию пептидных препаратов, и необходимость их перебора делает быстродействие вычислительных методов необходимым условием их применения в контексте разработки лекарств.
BiteNetPp доступен по ссылке: https://sites.skoltech.ru/imolecule/tools/bitenet/.
Источник информации и иллюстрации: Сколтех
























{kind=link}