

Ученые Санкт-Петербургского университета вместе с коллегами из Университета Цинхуа (Китай) будут разрабатывать новый высокоточный способ моделирования белковых структур — он может помочь создавать более эффективные лекарства и ответить на фундаментальные вопросы о работе белков. Проект исследователей получил грантовую поддержку Российского научного фонда (РНФ) и Государственного фонда естественных наук Китая (NSFC).
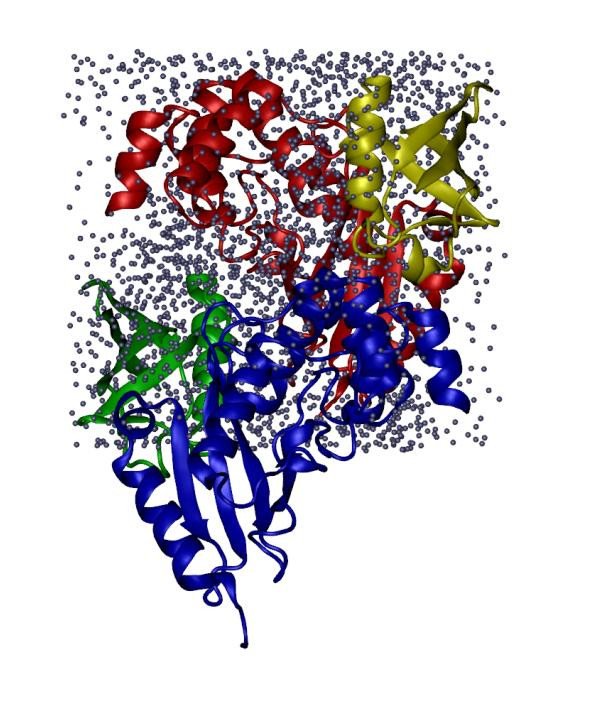
Полноатомная гибридная кристаллографическая/МД модель структуры белкового комплекса, растворенного в элементарной кристаллической ячейке
Представленные на конкурс проекты прошли независимую экспертизу как с российской, так и с китайской стороны. Победителями были признаны научные коллективы, получившие положительную оценку экспертов обеих стран. Исследования, проводимые в СПбГУ, будут получать поддержку от РНФ ежегодно на протяжении трех лет: с 2021 по 2023 год. Аналогичная по масштабу исследовательская программа в Университете Цинхуа будет финансироваться национальным научным агентством Китая NSFC.
Проект СПбГУ и Цинхуа посвящен разработке нового способа изучения белковых структур, который объединит уже используемые в науке методы исследования — рентгеновскую кристаллографию и молекулярно-динамическое (МД) моделирование. Для этих целей ученые создают программное решение, которое позволит более полно воссоздавать структуры белков, используя как статические характеристики, полученные с помощью рентгеновской дифракции, так и динамические, полученные методом молекулярной динамики. С российской стороны проект возглавит заведующий лабораторией биомолекулярного ЯМР СПбГУ профессор, PhD Николай Скрынников, с китайской — ведущий исследователь Университета Цинхуа, профессор, PhD Йи Шу.
«Методом рентгеновской кристаллографии сегодня воссозданы структуры множества белков, однако в этих структурах есть "слепые зоны", которые могут иметь принципиальное значение. К примеру, если у белка тело хорошо структурировано и имеет достаточно жесткую конструкцию, рентгеновские экспериментальные данные дают нам всю необходимую информацию и с высокой точностью позволяют реконструировать координаты и рассчитать его структуру. Но в то же время белковые молекулы имеют в своем составе подвижные элементы — например, петли — которые зачастую играют ключевую роль в функции белка. Например, подвижные петли могут служить своего рода "дверцей", которая контролирует доступ к активному сайту белка-фермента. И в том, что касается таких подвижных петель, рентгеновская кристаллография "пасует": она их либо не видит совсем, либо видит плохо. В таком случае недостающую информацию можно получить с помощью молекулярной динамики. И мы попытались создать гибридную методику, объединяющую оба эти подхода», — объясняет Николай Скрынников.
Полученные таким образом данные позволят усовершенствовать уже имеющиеся знания о структурах белков, что может помочь создателям медикаментов разрабатывать все более эффективные лекарства и находить более удачные подходы к лечению человека.
«Например, рациональный дизайн лекарственных средств осуществляется сегодня на основе кристаллографических структур. Развитие методов белковой инженерии также опирается на массив данных в мировом "банке данных" белков Protein Data Bank (PDB). Даже небольшое повышение точности и улучшение качества рентгеновских структур, подобное тому, которого нам удается добиться, будет способствовать дальнейшему прогрессу в этих областях», — говорит Николай Скрынников.
Первые шаги на пути к осуществлению замысла ученых были сделаны около 10 лет назад, когда Йи Шу был аспирантом у Николая Скрынникова в Университете Пердью (США). Однако довести идею до реализации удалось не сразу. Прогресс возник с появлением в команде исследователей выпускника СПбГУ, математика Олега Михайловского, который стал аспирантом Николая Скрынникова на химическом факультете Университета Пердью. Именно его междисциплинарные знания в области биологии, химии, биохимии, математики и программирования позволили создать программное обеспечение, представляющее собой синтез двух различных научных направлений. В качестве докторской диссертации Олег Михайловский под научным руководством Николая Скрынникова создал «надстройку» для пакета Amber — одного из двух крупнейших в мире наборов программ для биомолекулярного моделирования. Решение, созданное Олегом Михайловским, позволяет получать структуры белков, максимально полно используя при этом экспериментальные данные рентгеновской дифракции и обобщенную структурную информацию, содержащуюся в силовом поле молекулярной динамики. Теперь ученый работает под руководством Николая Скрынникова в лаборатории био-ЯМР СПбГУ.
«Проблема большинства белковых структур в том, что рентгеновские модели статичны, а белок — система динамическая. Некоторые части белков могут быть функционально важными, однако рентгеновская кристаллография не дает нам достаточного разрешения, чтобы их смоделировать. В этих местах модель структуры белка получается "замыленной", и непонятно, что в этой области происходит. Новый метод позволит смоделировать структуру белка более точно, опираясь как на данные рентгеновской кристаллографии, так и на возможности молекулярной динамики, чтобы понимать, как белок себя ведет в "пропущенных" на рентгеновской картинке элементах. Мы хотим использовать преимущества каждого из имеющихся способов, минимизировать недостатки и разработать новые методы уточнения белковых структур, используя эти две методики, чтобы получать более точные гибридные модели», — рассказывает разработчик программного обеспечения, научный сотрудник лаборатории био-ЯМР СПбГУ PhD Олег Михайловский.
Ученый добавил, что программные решения, над которыми он с коллегами работает в рамках проекта, воссоздают структуры белков на основе данных рентгеновской дифракции и МД-моделирования автоматически. Еще одно потенциальное преимущество этого способа — высокая скорость вычислений. «Структуры белков необходимы для работы ученым всего мира, а также представителям фармацевтической промышленности. На уточнение структур привычными способами уходит очень много времени — иногда процесс может достигать нескольких суток и более. Наш алгоритм требует всего нескольких часов вычислений, а в перспективе мы рассчитываем уменьшить это время до 10 минут. Кроме того, мы планируем расширить круг структур, которые можно оптимизировать с помощью разработанного нами протокола», — говорит Олег Михайловский.
Используя эти новые возможности, ученые планируют систематически уточнить значительную часть — десятки тысяч — белковых структур, содержащихся в мировом «банке данных» белков Protein Data Bank (PDB). Кроме того, исследователи намерены обеспечить широкий доступ к разработанной методике посредством сервиса ARX (Amber-based Refinement for protein Xtallography). В настоящее время созданный Олегом Михайловским и аспирантом лаборатории био-ЯМР Сергеем Измайловым ресурс функционирует в тестовом режиме на платформе СПбГУ. В планах научной группы — модернизация сервиса и расширение его возможностей, что позволит превратить его во всемирно востребованный инструмент для уточнения вновь получаемых рентгеновских структур.
Информация и фото предоставлены пресс-службой СПбГУ























{kind=link}