


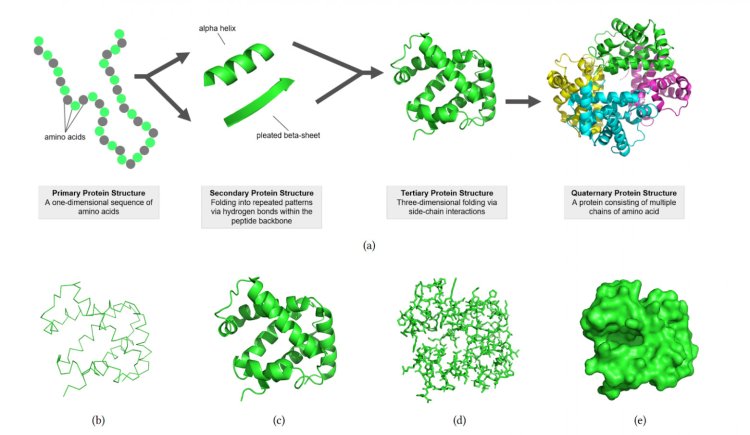
Белки называют кирпичиками жизни, хотя они не просто расходный материал, а «строители», «прорабы» и «архитекторы» в одном лице, обладающие тысячами разных специализаций и выполняющие несметное количество функций. В человеческом организме около 20 тыс. генов, и практически все они кодируют белки, каждый из которых состоит из уникальной аминокислотной последовательности. О вычислительном дизайне белков, их эволюции и процессе самопроизвольного сворачивания ― наш разговор с профессором Сколтеха Дмитрием Иванковым.
Справка: Дмитрий Николаевич Иванков ― кандидат физико-математических наук, биоинформатик, старший преподаватель и профессор Центра молекулярной и клеточной биологии Сколковского института науки и технологий. Научная группа под руководством Д.Н. Иванкова изучает эволюцию белков, их структуру, сворачивание и дизайн, используя широкомасштабный анализ данных, разработку алгоритмов, машинное обучение и молекулярное моделирование.
― В 2024 г. Нобелевская премия по химии была присуждена за предсказание трехмерной структуры белков, пространственного расположения их атомов, и вычислительный дизайн белков. Как развивалась эта область исследований?
― Пожалуй, точкой отсчета можно считать 1961 г., когда Кристиан Анфинсен провел эксперимент, за который в 1972 г. получил Нобелевскую премию по химии. Он экспериментально доказал, что трехмерная структура белка определяется исключительно его аминокислотной последовательностью в организме. На основе этого открытия возникло логичное предположение, что, зная ее, мы сможем быстро и эффективно предсказывать рабочую трехмерную структуру белков. С тех пор было представлено несколько различных подходов к тому, как это можно сделать, но ни один из них, к сожалению, не давал стопроцентной точности в таких предсказаниях. И лишь спустя более полувека, в 2020 г., в этой области случился долгожданный прорыв: ученые из компании Google DeepMind разработали вторую версию знаменитой программы AlphaFold, которая наконец смогла предсказывать трехмерную структуру белка с точностью, практически совпадающей с экспериментальными данными.
― Около 90%?
― Да. Увеличение этого показателя с 50% до почти 90% стало очень важным шагом в данной области знания. То, что за эту разработку рано или поздно дадут Нобелевскую премию, было ожидаемо.
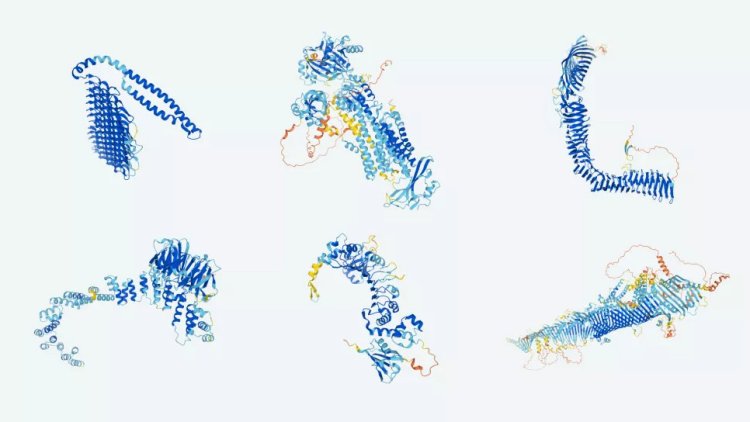
С помощью программы AlphaFold удалось раскрыть трехмерную структуру почти всех из 200 млн белков, известных науке. На фото: изображение белковых структур, предсказанных программой AlphaFold.
Фото: Google DeepMind
Другая часть Нобелевской премии по химии не менее интересна. Она была присуждена за вычислительный дизайн белков.
Над задачей рассчитать на компьютере белок с еще не наблюдавшейся в природе аминокислотной последовательностью ученые начали работать еще в 1980–1990-е гг. Значимых успехов начали достигать в конце 1990-х и начале 2000-х гг., в первую очередь в группе под руководством Дэвида Бейкера.
― Это что-то похожее на генерацию изображений нейросетью, но вместо картинок белки?
― Это подходящая аналогия. Если вы интересовались темой искусственного интеллекта, то наверняка знаете, что сначала он «разминается» на картинках с котиками и собачками, но затем наработанные усовершенствованные технологии переходят в другие области применения.
― Каков практический смысл генерации белков, которых нет в природе?
― Это имеет большое прикладное значение, например, для проведения реакций с участием белков-катализаторов. Если мы будем просто сидеть и ждать, пока какая-то химическая реакция завершится естественным образом, на это может уйти очень много времени.
Благодаря прогрессу в белковом дизайне у нас появилась возможность создавать принципиально новые белки, способные значительно ускорить протекание определенных реакций и получение конечного продукта.
Мы можем использовать такие белки, допустим, для разложения пластика. Это только одно из множества перспективных направлений.
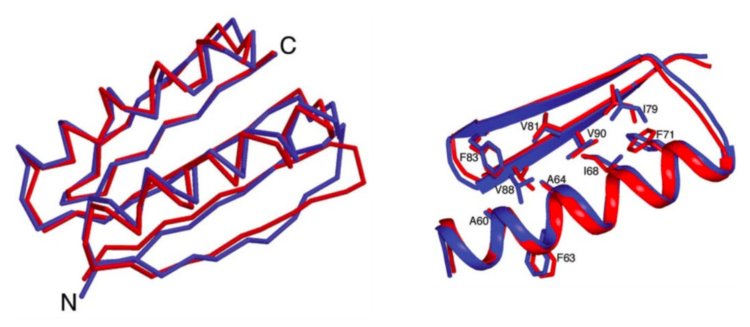
Полипептид Top7 — один из первых примеров белкового дизайна.
Фото: Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker / Science
― Насколько хорошо изучен процесс сворачивания белка в правильную трехмерную структуру?
― Достаточно хорошо. Ученые начали исследовать его еще в начале 1950-х гг. Первым белком, для которого смогли определить аминокислотную последовательность, был инсулин, а первые белки, для которых была определена трехмерная структура, ― миоглобин и гемоглобин. После этого одним из главных вопросов, волновавших ученых, стало сворачивание белков: как именно белок определяет конкретную структуру, в которую он свернется? Считалось, что раз эта структура рабочая, то она, соответственно, и самая стабильная. Но как белок ее находит? Человеком, который задался этим вопросом, был американский молекулярный биолог и программист Сайрус Левинталь. Кстати, он был одним из первых, кто визуализировал структуру белка ― прямо на экране осциллографа.
Согласно соображениям Сайруса Левинталя, белку для того, чтобы перебрать все возможные конформации, все допустимые взаимные расположения атомов и аминокислотных остатков, не хватит даже времени жизни Вселенной.
Но если он не переберет все возможные конфигурации, думал Левинталь, то не сможет найти самую стабильную структуру и свернуться в нее. Как же это происходит? Проблема получила название «парадокс Левинталя», и многие ученые пытались найти ее объяснение.
Сам Левинталь предположил, что белок сворачивается не в самую стабильную из всех структур, а в быстро доступную конформацию, отобранную в ходе эволюции. Позднее, в 1997 г., двое российских ученых, Азат Бадретдинов и Алексей Финкельштейн, показали в своей работе, что на самом деле к самой стабильной структуре автоматически существуют быстрые пути сворачивания, а параллельные пути только ускоряют процесс сворачивания.
Важно понимать, что в случае с белковыми структурами это происходит не потому, что эволюция постаралась (как предполагал Сайрус Левинталь), а потому, что это свойство белковой глобулы как таковой. И, конечно, среди множества путей сворачивания молекулы выбирают наиболее быстрый путь. Представьте, что перед вами работающий и сломанный эскалаторы. Уверен, что вы выберете тот, что сможет доставить вас из точки А в точку Б быстро и с минимальными усилиями. Так же и здесь. Только «эскалаторов» в данном случае намного больше, чем два. И следует отметить, что наличие сломанного эскалатора все же увеличивает число людей, доставленных из точки А в точку Б.
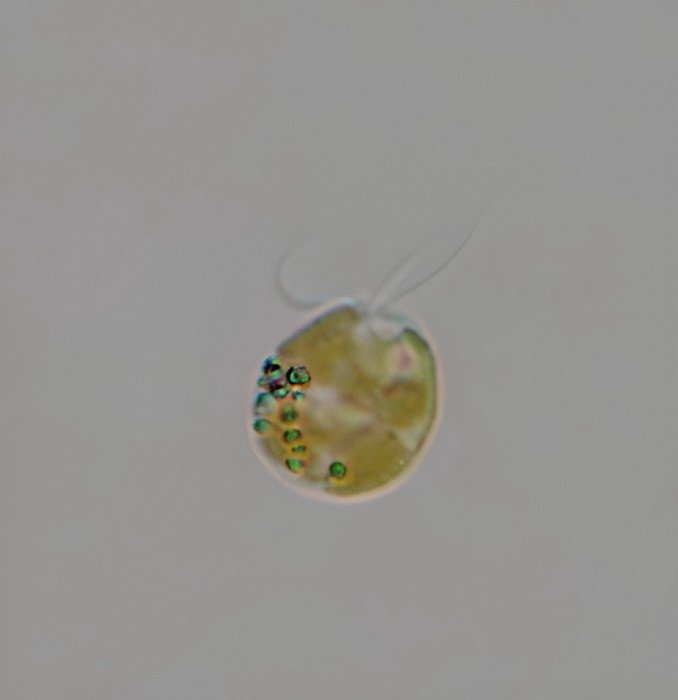
В 2024 г. в клетках водоросли Prymnesium parvum (на фото) был обнаружен самый большой из известных белков — PKZILLA-1. Его молекулярная масса достигает 4,7 МДа, а последовательность содержит более 45 тыс. аминокислотных остатков.
Фото: Greg Southard / Texas Parks and Wildlife Department
Таким образом, парадокс Левинталя был решен в 1997 г., и концептуально мы поняли, как сворачивается белок, но это не помогло нам предсказать его структуру. Далее последовали попытки сымитировать жизнь белка с помощью компьютера, посмотреть, как он свернется, но скорость работы компьютеров не настолько быстрая, как протекание аналогичных процессов в природе, и для того чтобы дождаться, пока белок свернется, нужно было очень много времени. Однако в 2010 г. появились первые работы, где очень-очень маленькие белки все-таки были свернуты, так что можно сказать, что в этой области был достигнут некий прогресс.
― Факторы извне влияют на этот процесс или все обусловлено сугубо внутренними причинами?
― Внешними условиями определяется только то, будет ли белок сворачиваться в трехмерную структуру или предпочтет оставаться в развернутом виде. Понятно, что существуют исключения, но они лишь подтверждают правило. Например, есть белки, у которых конечных конфигураций (рабочих структур) больше одной, то есть в таком случае часть молекул сворачиваются в одну конфигурацию, часть ― в другую, и переключение между ними, как правило, обусловлено как раз внешними факторами. Таких белков довольно мало: в 2008 г. их было известно с десяток, а сейчас, наверное, около сотни.
― Один из основателей молекулярной биологии Жак Моно в своей книге «Случайность и необходимость» писал о том, что появление жизни зиждется на способности белков распознавать другие молекулы, в том числе другие белки, по их форме. Что вы думаете об этом?
― Это очень интересная мысль, и я могу привести пару примеров, иллюстрирующих такое распознавание. В природе существует огромное количество белков, и все они разные. Если рассматривать белки класса ферментов, например алкогольдегидрогеназу (белок, расщепляющий этиловый спирт), то при благоприятных внешних условиях он сворачивается сам по себе (внешние условия, как я уже говорил, определяют лишь то, будет ли он сворачиваться или останется развернутым). То есть для формирования структуры этого белка не требуются никакие другие молекулы, которые он должен узнавать. Но есть другие белки, принадлежащие к классу нативно развернутых. Это значит, что они находятся в развернутом состоянии до тех пор, пока не найдется партнер, способный стабилизировать их трехмерную структуру. И если такой партнер находится, то, взаимодействуя с ним, им выгоднее свернуться, что они и делают.
― То есть они распознают его, а затем сворачиваются?
― Можно сказать и так. К такому классу белков относятся, например, почти все рибосомальные белки, которые при наличии рибосомы сворачиваются на ней, потому что взаимодействие с рибосомой делает их структуру рабочей, стабилизируя ее. В отсутствие этих взаимодействий они, в общем-то, предпочитают находиться в развернутом виде.
Возвращаясь к цитате Жака Моно. На этом примере мы видим, что существует класс белков, которые, не имея партнеров, сами по себе предпочитают быть развернутыми, но при появлении партнеров распознают их и предпочитают находиться в свернутом виде в комплексе с этими партнерами.
― Таких белков много или это единичные случаи? И что представляют собой их партнеры?
― Нет, это не единичные случаи. Зависит от того, что считать партнерами. Если относить к ним, например, молекулы воды, то получится, что такой партнер нужен абсолютно всем белкам, чтобы найти свою трехмерную структуру. Можно посмотреть на это с другой стороны: считать воду не партнером, а просто растворителем и сконцентрироваться на более специфических вещах, таких как взаимодействие белков с ионами металлов. Структуры некоторых белков стабилизируются взаимодействием с ионами металлов: цинка, железа и др. И в данном случае уже они выступают в роли партнеров.
Есть также белки, чья трехмерная структура стабилизируется какими-то более сложными молекулярными образованиями: к таким белкам относятся, например, гемоглобин и миоглобин, структура которых стабилизирована гемом.
Средний размер белка составляет около 200–400 аминокислот в цепочке (в зависимости от организма). На фото: фермент, взятый у бактерии Colwellia psychrerythraea.
Фото: Andrzej Joachimiak / Argonne National Laboratory / Flickr
― Сколько всего белков в человеческом организме?
― Тысячи. Практически всю работу в живых организмах выполняют именно они. Эта работа заключается во взаимодействии с какими-то другими атомными и молекулярными образованиями, часто ― с нуклеиновыми кислотами. В человеческом организме около 20 тыс. генов, кодирующих разные белки. Таким образом, в нашем организме существуют 20 тыс. типов белков. Если провести аналогию с фабрикой, то это 20 тыс. профессий. Каждый белок обладает уникальной аминокислотной последовательностью и сворачивается в трехмерную структуру, имеющую довольно жесткую форму: это позволяет белку выполнять предназначенную ему работу. Любой живой организм, по сути, представляет собой огромный конвейер или конвейеры с тысячами работников-белков, осуществляющих все химические реакции и другие жизненно важные процессы в организме.
Аминокислотная последовательность определяет структуру белка, а структура ― его функцию.
― В своих выступлениях вы говорили о том, что вам интересна эволюция белков. Исследования, которые проводятся в вашей лаборатории, как-то связаны с этой темой?
― Конечно. Сейчас мы сосредоточены на двух направлениях: исследовании стабильности белков и изучении их структуры и эволюции. Если говорить о первой теме, то очень хотелось бы, чтобы была разработана компьютерная программа, в которую можно было бы внести какую-то информацию о необходимых нам функциях белка, а затем, условно нажав на кнопку, получить на выходе аминокислотную последовательность такого белка, чтобы потом пойти в лабораторию и создать его. Это перспектива на будущее. Свои исследования в этой области, которая называется компьютерным дизайном белков, мы начали с самой простой задачи: изучения того, как можно предсказать изменение стабильности белка вследствие замены одной-единственной аминокислоты в его аминокислотной последовательности. Как ни странно, метода, предсказывающего, что будет в случае таких аминокислотных замен, до сих пор не существует.
― С помощью программы Rosetta Дэвида Бейкера это тоже нельзя сделать?
― Да. Более того, эта программа сильно уступает другим. Недавно мы изучали, как разные программы делают предсказания относительно изменения стабильности белка вследствие мутаций. Оказалось, они показывают корреляцию с экспериментальными данными на уровне от 35% до 60%, но это слишком мало. Только имея точность на уровне 90%, мы сможем сказать, что задача решена.
― Как именно вы пытаетесь решить ее в лаборатории?
― Разными способами. Так, например, мы разработали собственную программу, предсказывающую изменение стабильности белка вследствие мутации. Мы тренировались на новых данных, полученных в 2023 г. Котаро Цубоямой и другими учеными: коллеги опубликовали замечательный эксперимент, в котором для 850 тыс. различных вариантов разных белков были определены их стабильности в одной лаборатории за одну неделю.
― Это много?
― Это очень много по сравнению с тем, что было в нашем распоряжении раньше: около 13 тыс. белков, которые были определены в лабораториях мира, начиная еще с 1970-х гг. Благодаря новым данным 2023 г. мы смогли создать программу, показывающую корреляцию с экспериментом на уровне 75%. Но на этом нельзя останавливаться, и мы продолжаем свои исследования. Мы разработали свою программу ABYSSAL с помощью нейросетей, а также использовали в своих расчетах методы молекулярной динамики и квантовой химии.
Различные представления структуры белка.
Фото: Luiz Felipe Vecchietti, Minji Lee, Begench Hangeldiyev et al. / ArXiv.org
― Программой AlphaFold, о которой мы говорили ранее, пользуются ученые по всему миру. Как она повлияла на ваши исследования?
― Выход этой программы, безусловно, имел очень большое значение для нас. Часто можно услышать расхожее мнение о том, что эта программа решила все проблемы, касающиеся сворачивания белка. Но это не так. На самом деле этот процесс связан с множеством сложнейших вопросов, и предсказание трехмерной белковой структуры ― крайне важная задача, но далеко не единственная в этой области.
В 2022 г. мы первыми в мире опубликовали исследование, касающееся применимости структур, предсказанных AlphaFold, к программам, прогнозирующим изменение стабильности белков вследствие мутаций.
Нашей задачей было выяснить, хватает ли AlphaFold точности для того, чтобы мы могли пользоваться предсказанными ею структурами вместо того, чтобы брать их из банка белковых структур (потому что не для всех белков есть экспериментально определенная структура). В нашей работе было показано, что для хороших программ, предсказывающих изменение стабильности белка вследствие мутации по его по трехмерной структуре, можно смело использовать модели, произведенные AlphaFold, и потери точности не будет.
― Вернемся к эволюции белков. Какие задачи в этой области вам наиболее интересны?
― Нас очень интересует эволюция сама по себе. То есть не эволюция какого-то отдельного белка, а более фундаментальные вопросы. Один из них ― это так называемый эпистаз. Показать, что это такое, можно на примере любого функционального белка. Итак, у вас есть белок, который эволюционирует: в нем происходят мутации, которые закрепляются, допустим, в виде замены одной аминокислоты на другую в аминокислотной последовательности. Именно из-за таких замен мы видим в различных белковых организмах варианты одного и того же белка, отличающиеся по своей последовательности и даже немного по структуре. Но если эффект этой замены универсален и не зависит от замен, произведенных в этом белке ранее, то мы получаем полную предсказуемость! В таком случае мы могли бы экспериментально измерить все эффекты всех одиночных замен и затем для предсказания эффекта любого количества замен просто сложить эффекты этих одиночных замен.
― Но этого не происходит.
― Конечно, иначе мы бы получили полную предсказуемость фенотипа по генотипу. В действительности же оказывается, что эффект одной замены может зависеть и от других замен, ранее произведенных в этом белке. Это и есть эпистаз, и чтобы приблизиться к прогнозированию свойств белка, крайне важно изучать это явление.
Апогеем развития эволюционной биологии, на мой взгляд, могло бы стать появление компьютерной программы, в которую можно загрузить аминокислотную последовательность белка и на выходе получить информацию о том, что и как он делает в организме, то есть какова его «профессия» и какие партнеры ему нужны.
― Сколько таких аминокислотных замен может быть в одной белковой последовательности?
― Это очень интересная тема для обсуждения. Если мы будем делать такие замены случайно, наугад, то на один белок, состоящий в среднем из 200 аминокислотных остатков, хватит порядка пяти-шести замен, чтобы получить пятидесятипроцентную вероятность того, что он перестанет работать. Если же брать противоположный вариант, то есть убирать случайность и делать вполне определенную замену только на ту аминокислоту, которая встречается у этого же белка, но в других организмах, то выясняется, что в белке можно изменить до 70% всех аминокислотных остатков и при этом он продолжит выполнять свою функцию.
― То есть можно заменить аминокислотные остатки млекопитающих, например, рептильными — и белки все равно будут работать?
― Именно об этом я и говорю. Если сравнить один и тот же белок у людей, рептилий и, допустим, у грибов, то можно увидеть, что 70% аминокислотных остатков у этих протеинов ― разные, но при этом один и тот же белок и у нас, и у них выполняет одну и ту же функцию. И это очень интересно. Эти одиночные замены, которые мы с вами обсуждаем, очень удобны и полезны для изучения эволюции, но есть и другие типы замен — например, потеря целого куска гена, или, наоборот, вставка какого-то другого куска гена.
― Это тоже мутации?
― Да, это мутации, которые называются «делеции» и «вставки». При копировании наследственной информации во время деления клеток белок-фермент, осуществляющий это копирование, может делать какие-то ошибки.
― Не обусловленные воздействием внешней среды.
― Да. Вспомним аналогию, где наш организм выступает в роли конвейера, а белки ― это работники. На каждой стадии конвейера работает конкретный белок, и он, как и его коллеги по цеху, не застрахован от ошибок. Поэтому в организме человека за жизнь возникает от 20 до 100 новых мутаций, которые передаются следующему поколению.
― И чтобы лучше понять этот процесс, необходимо исследовать упомянутый вами эпистаз?
― В том числе, да. В нашей лаборатории мы разрабатываем специальные алгоритмы, позволяющие исследовать правила, характерные для эпистаза. Мы стремимся понять, как замена одного аминокислотного остатка в белковой последовательности связана с возможностью замен в других позициях белка. Я думаю, что исследование этого вопроса приблизит то время, когда мы научимся предсказывать фенотип по генотипу.












































{kind=link}